Recién Nacido Hipotérmico
Febrero 2019
Recien Nacido Hipotérmico
Joshua Erickson Department of Pediatrics, Eastern Virginia Medical School, Norfolk, VA
Presentación
Un neonato varón de 2 días nacido a las 38
semanas de una mujer con Gesta 1 , para 1 es traído al Servicio de
Urgencia debido a problemas de hipotermia, mala alimentación y disminución de
diuresis. Los resultados prenatales de laboratorio no son destacables , con
excepción de colonización por estreptococos del grupo B que se trató
adecuadamente con profilaxis con penicilina antes del parto. En evolución del
parto destaca meconio espeso por lo cual el niño se intuba y succiona con
extubación posterior. Apgar 4 y 9 a los 1 y 5 minutos, respectivamente. En
la nursery , se notó que el RN estaba amamantando bien y con deposiciones
normales . Fue dado de alta el día 2 después del nacimiento.
Después del alta, los padres se contactaron con el médico del RN para informar
que había disminuido el interés en la alimentación, la actividad disminuyó y
tenía temperatura rectal baja de 34.4 ° C. La familia recibió
instrucciones de llevar al neonato al Servicio de Urgencia para evaluación y
tratamiento adicionales. Al ingreso al Servicio de Urgencia el neonato tenía
temperatura 36.1 ° C y estaba decaído por lo cual fue llevado a un área de
reanimación. El examen físico inicial mostró un neonato de mal aspecto con
secreciones mucosas viscosas y reflejo de Moro exagerado. Se le administró un
bolo de solución salina normal de 10 ml / kg y luego otro bolo de solución
salina normal de 15 ml / kg.
Los hallazgos iniciales de laboratorio incluyen lo siguiente: glóbulos blancos 22.000 / ul ; hemoglobina 13.3 g / dL ; hematocrito 36.5%; recuento de plaquetas, 565.103 / ml ; neutrófilos 80%; bandas 1%; linfocitos 12%; monocitos 5%; sodio 154 mEq / L ; potasio 5.1 mEq / L ; cloruro 115 mEq / L ; bicarbonato 20 mEq / L nitrógeno ureico en sangre 6 mg / dL ; creatinina 1.4 mg / dL y glucosa 102 mg / dL .
Estos exámenes se interpretaron como leucocitosis con predominio neutrofílico pero sin una proporción significativa de células inmaduras, hipernatremia y anion gap acidosis metabólica.
Se tomaron muestras de líquido cefalorraquídeo (LCR) y orina y los resultados fueron los siguientes: glóbulos blancos de LCR de 7.000 / mL ; eritrocitos en LCR 149106 / mL (1491012 / L); glucosa LCR 63 mg / dL ; protein LCR 96mg / dL ; orina pH 6.0; esterasa leucocitaria negativa en orina; nitrito orina negativo; bacterias en orina muchas; leucocitos negativos. En este momento se consultó a Neonatología. Se recomendó el inicio de ampicilina y gentamicina y el ingreso en Neo Intermedio.
Posteriormente, el RN fue trasladado a la sala de cuidados especiales donde se observó que tenía un abdomen distendido; una radiografía abdominal anteroposterior mostró distensión intraluminal pero no aire libre, neumatosis ni gas venoso portal. Posteriormente, presentó apneas y requirió intubación y fue trasladado a UCIN para su posterior manejo.
Al ingreso a UCIN, se colocaron líneas arteriales y venosas umbilicales, a través de las cuales se administraron fluidos intravenosos a una velocidad de 140 ml / kg por día y se colocó sonda Foley para monitorizar diuresis. Dado el elevado nivel de creatinina, se suspendió la gentamicina y se inició cefotaxima. Tambien se inició tratamiento con oxacilina en este momento para ampliar la cobertura empírica. Se obtuvieron mediciones de gas de sangre capilar, nivel de amoniaco, hemoglobina / hematocrito repetidas y estudios de coagulación.
Los resultados de estas pruebas fueron los siguientes : pH de la sangre 7.07; PCO2 71 mm Hg ; exceso de base - 9; amoníaco más de 1.400 mg / dL , hemoglobina 9.9 g / dL ; hematocrito 29%; tiempo de protrombina 22.4 segundos; tiempo parcial de tromboplastina 46.6 segundos; Nivel de fibrinógeno 131.2 mg / dL .
Estos resultados se interpretaron como un trastorno metabólico probable con coagulopatía y acidosis respiratoria concomitante. Se administró al paciente 15 ml / kg de glóbulos rojos y 10 ml / kg de plasma fresco congelado.Los parámetros del ventilador se ajustaron para aumentar la ventilación y se consultó a genética médica. Con base en las recomendaciones genéticas, el neonato comenzó a recibir fenil-acetato de sodio como captador de nitrógeno por vía intravenosa e infusión de benzoato de sodio y arginina.
Al neonato se siguió realizando mediciones de amonio y lactato seriados; se realizaron estudios metabólicos de laboratorio adicionales, incluyendo aminoácidos plasmáticos, ácidos orgánicos en orina, perfil de acilcarnitina y ácido orótico en orina.
A pesar de los medicamentos de captación de nitrógeno, el paciente continuó teniendo niveles de amoníaco superiores a 1.358 mg / dL (970 mmol / L) durante el resto de la mañana de ingreso. Se colocó una línea venosa central de la vena yugular interna en preparación para la diálisis. Después de esto, el nivel de amonio del recien nacido comenzó a mostrar una tendencia a la baja a 393 mg / dL (281 mmol / L), continuó disminuyendo a 93 mg / dL (67 mmol / L) en las siguientes 24 horas y se normalizó al día siguiente.
Diagnóstico
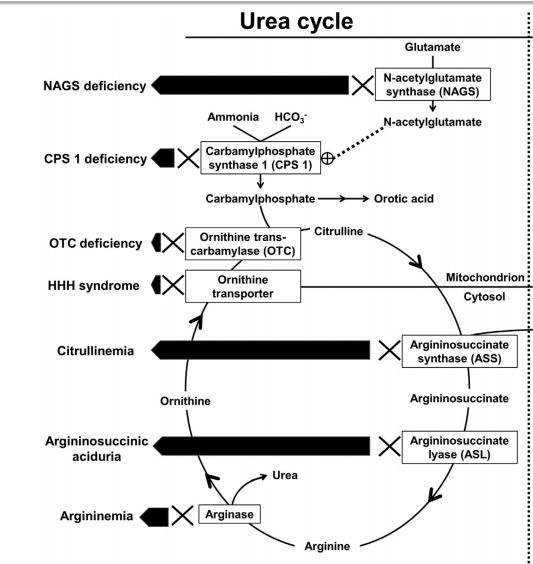
Los resultados de aminoácidos en plasma y ácidos orgánicos en orina se obtuvieron en 48 horas, y destacaba el aumento de citrulina, la excreción de anhídridos de argininosuccinato y el ácido orótico en orina positivo. Estos resultados sugieren un diagnóstico de deficiencia de argininosuccinate (ASA) liasa.
Se inició nutrición parenteral total con baja proteína el día 3 de hospitalización y las alimentaciones nasogástricas se iniciaron el día 6 de hospitalización con una mezcla de fórmula restringida de aminoácidos y leche materna, dado el diagnóstico presuntivo de deficiencia de ASA liasa. Las pruebas genéticas demostraron que el neonato era heterocigoto para 2 variantes patógenas del gen ASL, lo que confirma el diagnóstico.
La evaluación neonatal del recién nacido realizada 11 días después del nacimiento mostró un nivel elevado de citrulina. El R. nacido continuó mejorando y se le retiró todo el apoyo con excelentes alimentaciones orales. Posteriormente fue dado de alta del hospital con sonda nasogástrica en su lugar para medicamentos y para continuar con arginina, benzoato de sodio y fenilbutirato de sodio. Continuó recibiendo un seguimiento cercano por parte de su pediatra y genetista y, finalmente, recibió un trasplante de hígado ortotópico a los 8 meses de edad. Él sigue mejorando y logrando sus hitos de desarrollo.
La patología
La deficiencia de ASA liasa es una afección autosómica recesiva con una incidencia estimada de aproximadamente 1 de cada 70.000 nacidos vivos. (1)
ASA liasa es la enzima involucrada en el ciclo de la urea, que descompone el ASA para producir fumarato y arginina. La deficiencia o falta de esta enzima causa la acumulación de citrulina, el compuesto detectado en el screening neonatal para la identificación de la deficiencia de ASA liasa.
La citrulina elevada también se puede observar en Citrulinemia, otro trastorno del ciclo de la urea, así como tambien en la deficiencia de piruvato deshidrogenasa. Como tales, los aminoácidos plasmáticos deben ser chequeados para determinar los niveles de ASA y sus anhídridos para confirmar el diagnóstico de deficiencia de ASA liasa.
Los neonatos con deficiencia de ASA liasa (u otros trastornos del ciclo de la urea) presentarán típicamente dificultades de alimentación e hipotermia después de un período inicial de bienestar. Sin embargo, a medida que aumentan los niveles de amonio , el RN tendrá un aumento de signos del sistema nervioso central debido a edema cerebral, y puede incluir alcalosis respiratoria, letargo, convulsiones y coma.
Únicos para deficiencia de ASA liasa son
los hallazgos de hipertensión sistémica y tricorrexis nodosa (áreas de alopecia
parcial con cabello quebradizo). (2)
Por lo general, se trata en la fase aguda con terapia de captación (scavenger)
de nitrógeno, suplementos de arginina y dieta libre de proteínas, a la vez que
proporciona la glucosa adecuada para prevenir el catabolismo. Si el médico no
puede normalizar los niveles séricos de amonio, debe considerarse la
hemodiálisis. A largo plazo, estos pacientes se benefician del trasplante
ortotópico de hígado porque esto evitará más crisis metabólicas.
Ciclo de la Urea
Lecciones para el Clínico
Los médicos deben tener un alto índice de sospecha de hiperamonemia cuando los neonatos presentan hipotermia, taquipnea, alimentación deficiente y letargo entre los 2 y 7 días de edad.
Una vez que se realiza el diagnóstico de hiperamonemia, la terapia rápida con arginina, terapia de captadores de nitrógeno y prevención del catabolismo, a la vez que restricción de ingesta de proteínas, es crucial para reducir los niveles de amoníaco y prevenir potencialmente el impacto en la neurocognición.
Una elevación de la citrulina en el screening neonatal es sugerente de deficiencia de argininosuccinato liasa, citrulinemia o deficiencia de piruvato carboxilasa.
Para aclarar el diagnóstico a partir de este punto, es necesario un panel de aminoácidos del suero.
Especificaciones del contenido de American Board of Pediatrics Neonatal - Perinatal
Conocer las causas y diagnóstico diferencial de la encefalopatía metabólica.
Conocer las manifestaciones clínicas, laboratorio y el tratamiento de trastornos del metabolismo del ciclo de la urea.
Reconocer las manifestaciones clínicas y de laboratorio de acidosis metabólica y alcalosis metabólica en neonatos.
Conocer las causas y diagnóstico diferencial de acidosis metabólica y alcalosis metabólica en neonatos.
Referencias
Nagamani SCS, Erez A, Lee B. Argininosuccinate lyase deficiency. GeneReviews. https://www.ncbi.nlm.nih.gov/books/NBK51784/. Accessed November 5, 2018
Fichtel JC, Richards JA, Davis LS. Trichorrhexis nodosa secondary to argininosuccinicaciduria. Pediatr Dermatol. 2007;24(1):25–27
Suggested Readings
Brusilow S, Horwich A. Urea cycle enzymes. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001; chap 85:1909–1963
Robberecht E, Maesen S, Jonckheere A, Van Biervliet S, Carton D. Successful liver transplantation for argininosuccinate lyase deficiency (ASLD). J Inherit Metab Dis. 2006;29(1): 184–185