Púrpura de Schönlein-Henoch, enfermedad de Kawasaki y otras vasculitis
M.S. Camacho Lovillo, M.J. Lirola Cruz* Hospital Virgen del Rocío de Sevilla. *Instituto Hispalense de Pediatría
Pediatr Integral 2013; XVII(1): 34-46
El término vasculitis se refiere a la
inflamación y necrosis de los vasos sanguíneos. Se suelen afectar varios órganos
o sistemas, con unas manifestaciones clínicas dependientes de la localización y
tamaño de los vasos implicados. Pueden ser primarias o secundarias a
infecciones, tumores, drogas u otras enfermedades reumáticas.
Su frecuencia es muy baja en niños, a excepción de la Púrpura de Schönlein-
Henoch y la enfermedad de Kawasaki. Debido a la afectación multisistémica y la
baja incidencia, el diagnóstico es con frecuencia difícil y,
consecuentemente, tardío, lo cual suele asociarse a una importante morbi-mortalidad(1,2).
En el año 2005, la Liga Europea contra el Reumatismo (EULAR) y la Sociedad
Europea de Reumatología Pediátrica (PRES) desarrollaron la primera clasificación
pediátrica de vasculitis (Tabla I) (3).
Tabla I. Clasificación de la vasculitis en niños
-
Vasculitis predominantemente de grandes vasos
-
Arteritis de Takayasu
-
-
Vasculitis predominantemente de vasos de mediano calibre
-
Poliarteritis nodosa infantil
-
Poliarteritis cutánea
-
Enfermedad de Kawasaki
-
-
Vasculitis de predominio en pequeños vasos
-
Granulomatosas
-
Granulomatosis de Wegener
-
Enfermedad de Churg-Strauss
-
-
No granulomatosas
-
Poliangeítis microscópica
-
Púrpura de Schönlein-Henoch
-
Vasculitis leucocitoclástica exclusivamente cutánea
-
Vasculitis urticariforme hipocomplementémica
-
-
-
Otras vasculitis
-
Enfermedad de Bechet
-
Vasculitis secundaria a infección, enfermedades malignas y drogas
-
Vasculitis asociada con conectivopatías
-
Vasculitis del sistema nervioso central
-
Síndrome de Cogan
-
No clasificadas
-
Los hallazgos clínicos y de laboratorio que deben hacer sospechar la existencia de una vasculitis se reflejan en la tabla II.
Tabla II. Características sugestivas de vasculitis
-
Fiebre prolongada de origen desconocido
-
Lesiones cutáneas sugestivas (púrpura palpable, gangrena, nódulos dolorosos, livedo reticularis…)
-
Neuropatía periférica de causa desconocida
-
Artralgias, artritis, miositis, serositis
-
Enfermedades renales, pulmonares o cardiovasculares de causa no determinada, cuando hay afectación multisistémica
-
Parámetros de laboratorio indicativos de inflamación: leucocitosis, aumento de VSG/PCR, eosinofilia, hipocomplementemia, crioglobulinemia, inmunocomplejos circulantes…
Las pruebas de imagen son útiles, sobre todo en las vasculitis de mediano y grandes vasos, siendo en la mayoría de casos necesaria la biopsia de tejidos afectos. El tratamiento generalmente se realiza con corticoides e inmunosupresores y se requiere un abordaje multidisciplinar. El limitado número de estudios específicos en niños condiciona que muchos aspectos sobre su manejo se extrapolen a partir de la experiencia publicada en adultos(1,2).
Púrpura de Schönlein-Henoch
Introducción
La púrpura de Shönlein-Henoch (PSH) es la vasculitis sistémica más frecuente en
los niños. El primer caso fue descrito por Willian Heberden en 1801 en un niño
de 5 años con erupción purpúrica, hematuria macroscópica, dolor abdominal,
deposiciones sanguinolentas, vómitos, artralgias y edema. En 1837, Johann
Schönlein añadió el componente articular y denominó a esta entidad “peliosis
reumática” o “púrpura rubra” y, algo más tarde, Eduard Heinrish Henoch, alumno
de Schönlein, completó su descripción.
La naturaleza vasculítica de la PSH, o púrpura anafilactoide, fue descrita por
Gairdner en 1948. Sus manifestaciones clínicas más frecuentes son bien conocidas
: púrpura palpable, artritis, dolor abdominal, sangrado intestinal y nefritis,
aunque cualquier órgano puede verse afectado. Son consecuencia de una vasculitis
leucocitoclástica de pequeños vasos debida al depósito de IgA1 en la pared de
los vasos y del mesangio renal.
Se ha asociado a una gran variedad de microorganismos, drogas y otros agentes
ambientales. En general, su curso es autolimitado aunque el grado de afectación
renal condicionará su pronóstico a largo plazo (1,4-6).
Epidemiología
La PSH puede aparecer en todos los grupos de edad, siendo más frecuente durante
la infancia (entre los 3 y 15 años), ocurriendo el 50% de los casos en menores
de 5 años y el 75-90%, en menores de 10 años. Los hallazgos clínicos son, con
frecuencia, atípicos en las edades extremas y de mayor gravedad en el adulto;
sin embargo, en los menores de 2 años son menos probables las complicaciones
renales y gastrointestinales.
La incidencia oscila entre los 10 y 20(4). casos por cada 100.000 niños, pudiendo alcanzar los 70,3 casos/ 100.000 en el grupo de edad entre los 4 y 7 años(4). La distribución según el sexo es similar, aunque con predominio en varones en algunas series (1,5- 2:1). No existe un claro predominio racial, los afroamericanos rara vez se afectan. La enfermedad es más frecuente en invierno, otoño y primavera, lo que hace probable la implicación de determinados procesos infecciosos en su patogénesis.
Etiopatogenia
Las investigaciones tempranas sobre susceptibilidad genética de la PSH se
focalizaron en las asociaciones HLA. Estudios procedentes de España, Italia
y Turquía, informaron sobre la asociación de los alelos HLA-DRB1*01, HLA DRB1*11
y HLA- DRB1*14 con esta enfermedad. Un estudio reciente de Turquía mostraba que
el alelo HLA-B35 se asociaba con un riesgo elevado de PSH. Un estudio anterior
español informaba sobre la asociación del HLA-B35 con el
aumento de riesgo de nefritis, pero no sobre el riesgo total de PSH(5).
La PSH aparece hasta en el 7% de los pacientes con fiebre mediterránea familiar
(FMF), enfermedad autoinflamatoria determinada genéticamente. Estudios
procedentes de Israel y Turquía muestran un aumento significativo de la
prevalencia de mutaciones que afectan al gen MEFV, causa de la FMF, en los niños
con PSH en comparación con la población general. Es improbable que estos
hallazgos sean extensibles a otros países en los que la FMF es mucho menos
común(5).
Se han estudiado polimorfismos en genes que codifican citocinas proinflamatorias,
moléculas de adhesión celular y moléculas asociadas con la activación de células
endoteliales, focalizándose la mayoría de los trabajos en el TNF-alfa, IL-1beta,
IL-8, TGB-beta y VEGF. No se ha llegado hasta la fecha a conclusiones firmes
respecto a su asociación genética con la PSH(5).
La patogénesis de la PSH continúa siendo desconocida; sin embargo, en general,
se piensa que es una enfermedad mediada por el depósito de inmunocomplejos,
caracterizados por la presencia de IgA1 polimérica, predominantemente a nivel de
los capilares dérmicos, gastrointestinales y glomerulares. Los depósitos
granulares patognomónicos de IgA y C3 en el mesangio son indistinguibles de los
que se observan en la nefropatía por IgA.
Existen 2 subclases de IgA, la IgA1 y la IgA2. La IgA1 contiene una región
bisagra con múltiples lugares de glicosilación. La glicosilación aberrante de
esta región de la IgA1 podría explicar su participación exclusiva en la
patogénesis de la PSH. El aumento de la producción de IgA polimérica por parte
del sistema inmune de las mucosas en respuesta a la presentación de antígenos,
tales como bacterias, virus u hongos, se contempla como posible mecanismo
desencadenante de la PSH. Se ha documentado la existencia de niveles séricos
elevados de IgA1, complejos inmunes que contienen IgA1 (de pequeño peso
molecular), IgA- ANCA, IgA- FR en los pacientes con PSH. En aquellos que,
además, presentan nefritis (PSHN), se detectan complejos inmunes circulantes
IgA1-IgG de gran masa molecular. Así mismo, los niveles séricos de de IgA1 con
defecto de galactosa (Gd-IgA1) son significativamente más elevados en los
pacientes con PSHN que en los controles sanos y en los pacientes con PSH sin
nefritis.
Recientemente, se ha demostrado que los niveles en sangre y en orina de
leucotrieno B4 (LTB4), potente activador de los neutrófilos e inductor de su
quimiotaxis, son más altos en los pacientes con PSH y nefritis. Mientras
que, los niveles de lipoxina A4 (LXA4), inhibidora de la activación y
reclutamiento de los neutrófilos e inhibidora de la producción de citoquinas
proinflamatorias, son más bajos. Estos hallazgos pueden ayudar a explicar el
papel prominente de los neutrófilos en la patogénesis de la PSH (5).
Manifestaciones clínicas
Manifestaciones cutáneas
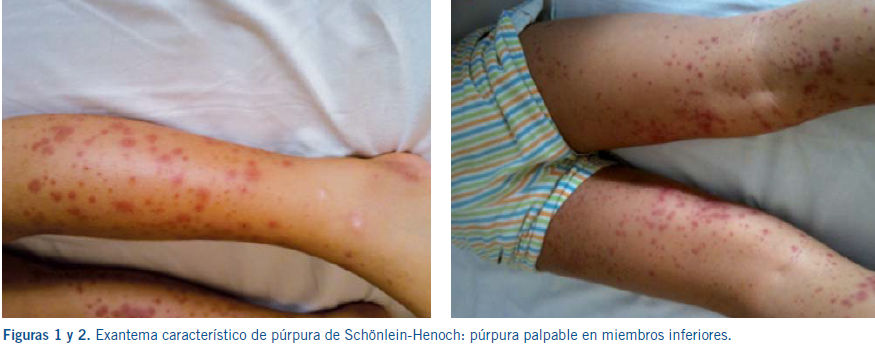
La lesión cutánea característica es la púrpura palpable, presentando el paciente
desde petequias a grandes equimosis, pudiendo estar precedidas de un exantema
maculopapular eritematoso o urticarial. Aparecen de forma simétrica en las zonas
declive (miembros inferiores y nalgas) (Figuras.
1 y 2), aunque también pueden
encontrarse en los brazos, cara, orejas y espalda. Al inicio del cuadro, puede
acompañarse de edema de cuero cabelludo, cara, manos, pies y escroto,
sobre todo en niños pequeños. Las lesiones ampollosas o hemorrágicas
y necróticas son raras en los niños (2%), ocurriendo hasta en el 60% del
paciente adulto.
Manifestaciones digestivas
Se describen en el 50-75% de los pacientes, siendo el primer síntoma de la
enfermedad en el 14-36% de los casos. Se producen como consecuencia del edema y
la hemorragia secundaria a la vasculitis de la pared intestinal. El síntoma más
frecuente es el dolor abdominal de tipo cólico. Otras manifestaciones pueden
ser: hemorragia digestiva (masiva en el 2%), invaginación intestinal, úlceras,
perforaciones, pancreatitis aguda, hydrops de la vesícula biliar y enteropatía
pierde proteínas.
Manifestaciones articulares
La artritis o artralgia puede ser el primer síntoma de la enfermedad en el
15-25% de los pacientes, encontrándose algún grado de afectación articular en el
82% de los casos. Característicamente, la inflamación es periarticular,
dolorosa, sin eritema ni calor pero con limitación, afectando con mayor
frecuencia a las grandes articulaciones de miembros inferiores. Son transitorias
y se resuelven en pocos días sin dejar deformidad.
Manifestaciones renales
Se producen en el 20-50% de los
pacientes y es el factor pronóstico más importante de la enfermedad. La
afectación renal se manifestará con: hematuria microscópica/macroscópica,
proteinuria, síndrome nefrótico/nefrítico, fracaso renal e hipertensión, siendo
la afectación severa en el 5-7% de los casos. Se desarrollará durante el 1º mes
de la enfermedad en el 75-80% de los pacientes, y en el 97-100% de los casos a
los 3 meses de inicio de la enfermedad. Se han descrito pocos casos en los que
la afectación renal se desarrolló varios años después de la presentación de la
enfermedad.
Manifestaciones neurológicas
Son raras, aunque la cefalea seguida
de una ligera encefalopatía con mínimos cambios en el estado mental, tales como:
labilidad emocional, apatía e hiperactividad, podría ser más frecuente de lo que
se pensaba. Podemos encontrar alteraciones electroencefalográficas y
convulsiones. Se han descrito casos de hematoma subdural, hemorragia
subaracnoidea, hemorragia cerebelosa, sangrado intraparenquimatoso e infarto.
Manifestaciones pulmonares
La afectación pulmonar es rara. Aunque ocurre con mayor frecuencia en el adulto,
se han descrito casos aislados en los niños de hemorragia difusa alveolar,
neumonía intersticial y fibrosis intersticial.
Manifestaciones urológicas
La orquitis es un hallazgo relativamente común en el niño con PSH, encontrándose hasta en el 24% de los casos. Puede llegar a ser necesaria la exploración quirúrgica para descartar la existencia de torsión testicular(1,4-6).
Diagnóstico
No existen pruebas de laboratorio específicas para el diagnóstico de la
enfermedad ; por lo que, nos basaremos, fundamentalmente, en los hallazgos
clínicos, precisándose en ocasiones hallazgos anatomopatológicos(5,7) (Tabla III).
Tabla III.- Criterios de clasificación de la púrpura de Schönlein-Henoch
-
Púrpura palpable en presencia de al menos uno de los siguientes hallazgos:
-
Dolor abdominal difuso
-
Artritis (aguda) o artralgia
-
Afectación renal (hematuria y/o proteinuria)
-
Biopsia mostrando los depósitos de IgA
-
La investigación irá encaminada a
descartar otros posibles diagnósticos y a conocer la extensión de la afectación
orgánica. En el estudio inicial se incluirán : hemograma,
coagulación, creatinina, urea e iones, perfil hepático y óseo, uroanálisis para
detectar hematuria y proteinuria y, si fuese necesario, determinación de sangre
oculta en heces. Si se identifica proteinuria en la tira reactiva, se
determinará en orina de la mañana el índice proteína: creatinina. Si el
diagnóstico fuese dudoso, añadiríamos un perfil autoinmune completo, incluyendo:
ANA, antiDNAds, ANCA, inmunoglobulinas, C3 y C4. Podremos encontrar anemia,
leucocitosis, un discreto aumento de la VSG y, en algunos casos, una función
renal y/o hepática alteradas; en pacientes con proteinuria importante, podemos
encontrar hipoalbuminemia.
La trombocitosis se ha asociado con enfermedad más severa. Los estudios
estándares de coagulación son habitualmente normales, aunque la actividad del
factor XIII se encuentra disminuida en relación con enfermedad más severa; no se
aconseja su determinación rutinaria. La IgA se encuentra elevada en la mitad de
los pacientes y no se correlaciona con la severidad de la enfermedad; pueden
encontrarse inmunocomplejos circulantes de IgA.
El significado de la presencia de IgA ANCA en la PSH está aún en discusión. En
general, el estudio básico inmunológico suele ser normal, encontrándose en
alguna ocasión niveles descendidos de C3 y C4. En caso de existir sospecha de
proceso infeccioso, en función de los síntomas encontrados, realizaríamos :
hemocultivo, cultivo faringe, urocultivo y Rx de tórax. Si existe evidencia de
infección estreptocócica reciente, determinaríamos los títulos de ASLO y antiDNA
asa B. La investigación viral podría revelarnos la causa precipitante de la
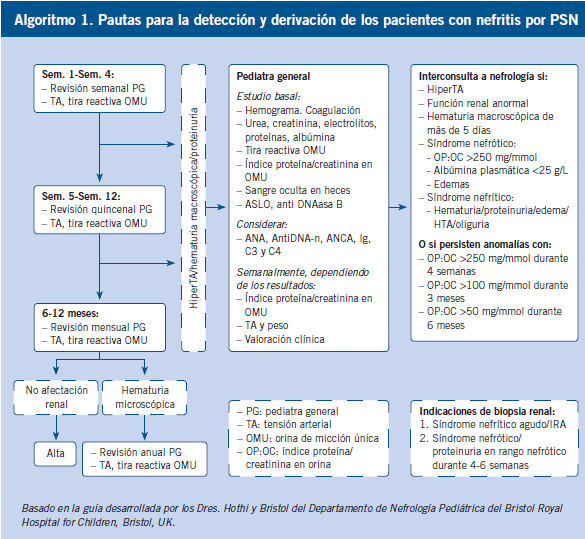
enfermedad. Si existe afectación renal, se recomienda realizar los controles y
pruebas indicados en el
Algoritmo 1.
La biopsia cutánea, si se realizase (normalmente ante una presentación atípica y dudas diagnósticas), mostraría una vasculitis leucocitoclástica de pequeños vasos con depósitos de IgA e infiltración de neutrófilos y células mononucleares perivasculares. En la biopsia renal, podemos encontrar desde glomerulonefritis con lesiones focales y/o segmentarías hasta la formación de semilunas. También, encontraremos depósitos de IgA en la inmunofluorescencia. La ecografía abdominal puede mostrar un engrosamiento de las paredes del intestino delgado y grueso, ayudará a descartar la existencia de una invaginación intestinal. Realizaremos ecografía escrotal en los casos de escroto agudo para descartar la existencia de una torsión testicular(1,4-7).
Tratamiento
Afectación cutánea
El reposo disminuye la aparición de nuevas lesiones cutáneas, no precisando, en
general, tratamiento específico añadido. En caso de lesiones bullosas,
existen notificaciones del éxito del tratamiento con corticoides. Se han
utilizado también agentes ahorradores de corticoides, como la dapsona y la
colchicina.
Afectación articular
Normalmente responden a tratamiento con antiinflamatorios no esteroideos, aunque
existen datos sobre la rápida respuesta y el acortamiento de la duración de los
síntomas con el uso de corticoides.
Enfermedad gastrointestinal
El uso de prednisolona a 1-2 mg/kg (máximo, 60 mg) se podría considerar en niños
con PSH y dolor abdominal moderado-severo, una vez descartada patología
abdominal significativa, como la invaginación. En caso de vasculitis
gastrointestinal muy severa (enteropatía pierde-proteínas y la hemorragia
gastrointestinal severa, entre otras), se ha descrito el éxito del tratamiento
con infusión de gammaglobulinas, pulsos de metilprednisolona y plasmaféresis. El
dolor abdominal persistente o crónico es poco común, pero parece responder a
metotrexate o micofenolato mofetilo, aunque deben ser valorados sus efectos
secundarios gastrointestinales.
Enfermedad renal
El tratamiento de la PSHN sigue siendo controvertido, no existiendo
evidencia suficiente sobre la mejor guía de tratamiento para la PSHN
establecida. Se proponen tratamientos con: prednisolona, metilprednisolona,
ciclofosfamida, azatioprina, ciclosporina A, micofenolato mofetilo, dipiridamol,
warfarina, plasmaféresis y rituximab. Los efectos antihipertensivos y
renoprotectores de los inhibidores de la enzima convertidora de la angiotensina
(IECA) o de los antagonistas del receptor de la angiotensina II (ARA II) están
bien documentados en el adulto con hipertensión y/o insuficiencia renal crónica.
En un estudio retrospectivo más reciente, aportan su experiencia positiva con el
tratamiento de la PSH con nefropatía severa y la nefropatía IgA con la
combinación de esteroides, ciclofosfamida, IECA y ARA II, consiguiendo una
respuesta adecuada en el 54% de los niños con cambios histológicos severos (>
estadio III) en la biopsia inicial(8-11).
Pronóstico
La PSH es generalmente una enfermedad autolimitada (en 2-4 semanas), aunque
hasta el 33% de los pacientes pueden presentar síntomas recurrentes (entre 1 y 6
episodios). Estas recurrencias suelen acontecer durante los 2-3 primeros meses,
aunque se describen recaídas que sobrepasan los 18 meses del inicio de la
enfermedad. Normalmente, los síntomas son similares a los del debut y parece ser
que aquellos pacientes con afectación renal pueden recaer con mayor facilidad.
El pronóstico a largo plazo de los niños con PSH se relaciona predominantemente
con la existencia de enfermedad renal. Aunque ningún hallazgo es absolutamente
predictivo, muchos estudios coinciden en que la presencia de síndrome
nefrítico/ nefrótico, la disminución de la actividad del factor XIII, la
hipertensión, el desarrollo del fallo renal al inicio de la enfermedad y la
presencia de esclerosis glomerular/semilunas/afectación tubulointersticial
(lesiones histopatológicas clase IV y V), se definen como factores de mal
pronóstico; de tal manera que, aunque puedan presentar una recuperación inicial,
en el seguimiento a largo plazo (más de 20 años en algunos casos), casi la mitad
de estos pacientes pueden presentar hipertensión o insuficiencia renal. Aquellas
mujeres a las que se les diagnosticó una PSH durante la infancia, presentarán
con mayor frecuencia hipertensión arterial y proteinuria durante el embarazo. El
tratamiento inicial de la PSH con corticoides no previene el desarrollo de
nefritis(6). Por todo lo referido anteriormente, dado el potencial riesgo de
deterioro renal de los pacientes con historia de PSHN, se aconseja su
seguimiento de por vida. En aquellos pacientes sin alteraciones del sedimento
urinario y con tensiones arteriales normales, el seguimiento se podría abandonar
a los 6-12 meses del inicio de la enfermedad o de la última recaída.
Enfermedad de Kawasaki
Introducción
Fue descrita por primera vez por Tomisaku Kawasaki en Japón en 1967. Desde
entonces su incidencia ha ido en aumento, describiéndose en todos los grupos
raciales y étnicos. Su etiología es aún desconocida. Su importancia se debe a
que el 15-25% de los niños no tratados desarrollan anomalías coronarias (AC) que
puede conducir a infarto de miocardio, muerte súbita o enfermedad isquémica
cardiaca. El tratamiento va dirigido a reducir la inflamación y prevenir el
desarrollo de AC(12,13).
Epidemiología
La EK afecta principalmente a niños entre 6 meses y 5 años, existiendo una mayor
proporción de varones (1,5:1). La incidencia más alta se ha registrado en Japón
(13). En nuestro país se estima que la incidencia anual acumulada es similar a
la encontrada en Estados Unidos (20,8/100.000 niños menores de 5 años en 2006)
(12). Durante el periodo 1998-2009, la incidencia en Noruega, Suecia y Finlandia
fue de 5,4, 7,4 y 11,4, respectivamente; mientras que, en ese periodo en Japón,
la incidencia aumentó de 111,7 a 218,6/100.000 niños menores de 5 años. Se
desconocen las causas de este incremento, si bien ha contribuido un mejor
reconocimiento y diagnóstico de la enfermedad(14). Las recurrencias son poco
frecuentes (Japón 3% y Estados Unidos 1-2%). Los casos familiares son raros
(0,7-2,1%) y el 50% aparecen en los 10 días siguientes al primer caso. En Japón
existe un predominio de la enfermedad en los meses de invierno y primavera; si
bien,
el predominio estacional varía de unos países a otros(15).
Etiopatogenia
La EK se debe a una respuesta inmunológica
inapropiada a uno o más agentes infecciosos que actuarían como desencadenantes
en individuos genéticamente
susceptibles (Tabla
IV)
(16,17).
Tabla IV. Etiología de enfermedad de Kawasaki
-
Sugieren un origen infeccioso
-
Manifestaciones similares a enfermedades exantemáticas pediátricas
-
Curso clínico autolimitado y, habitualmente, no recurrente
-
Estacionalidad
-
Existencia de endemias y epidemias
-
Propagación en forma de ondas durante epidemias
-
Limitada casi exclusivamente a edad pediátrica
-
Baja incidencia en 3 primeros meses de edad (¿protección por anticuerpos maternos?)
-
Hallazgos de laboratorio sugestivos de infección
-
-
Sugieren factores genéticos
-
Mayor incidencia en algunos países como Japón
-
Incidencia en niños de origen japonés tan elevada como en su país de origen
-
Existencia de casos familiares no contemporáneos
-
Riesgo de aparición en gemelos del 13%
-
La hipótesis que relaciona la EK con determinadas toxinas bacterianas
estreptocócicas o estafilocócicas que actuarían como superantígenos es, hoy día,
controvertida. El aumento de IgA en el tracto respiratorio que se detecta en
pacientes con EK sugiere una puerta de entrada respiratoria del agente
etiológico. El coronavirus New Haven ha sido identificado en secreciones
respiratorias de niños con EK, así como el coronavirus y bocavirus(12,16).
Actualmente, existen múltiples estudios encaminados a identificar marcadores
genéticos de susceptibilidad de la enfermedad, de severidad y de resistencia al
tratamiento [CTL-4, caspasa 3, IL- 10, IL-1B, CD40L, PD-1, ORAI1 e inositol
1,4,5 trifosfato3kinasaC(ITPKC)](12).
El paso que conduce a la arteritis
coronaria aún no está aclarado, si bien la activación de células endoteliales,
monocitos/macrófagos CD68, linfocitos CD8 y células plasmáticas IgA monoclonales
parece estar implicada. Un trigger infeccioso aumentaría la producción de
citocinas, como: TNF-α, IL 1 e IL 6, que inducirían nuevos antígenos
endoteliales y se generarían anticuerpos contra ellos. Se produce un infiltrado
de macrófagos y linfocitos en la pared arterial que secretan mediadores
inflamatorios y enzimas que contribuirían al daño vascular. La inflamación acaba
produciendo una destrucción de la media y la formación de aneurismas. En los
estadios precoces, se produce una infiltración de neutrófilos que producirían
óxido nítrico. Las células T reguladoras (Tregs) están disminuidas en sangre
periférica en EK aguda y aumentados los Th17(16,17). Se suelen afectar las
arterias de tamaño medio extraparenquimatosas (celíacas,
mesentéricas, femoral, ilíaca, renal, axilar y braquial) y, en especial, las
arterias coronarias. Los aneurismas son más frecuentes en las zonas proximales y
en las bifurcaciones, ya que son las zonas de mayor estrés de la pared arterial.
El enlentecimiento del flujo en la zona dilatada favorece la formación de
trombos. Las coronarias que se afectan con mayor frecuencia son la arteria
coronaria izquierda, la descendente anterior y la coronaria derecha(13,16).
Clínica
En ausencia de una prueba diagnóstica específica o características clínicas patognomónicas, se han establecido unos criterios clínicos para ayudar al diagnóstico de EK (Tabla V) (13).
Tabla V. Criterios clínicos de enfermedad de Kawasaki *
-
Fiebre persistente ≥5 días
-
Presencia de al menos 4 características principales :
-
Cambios en extremidades
-
Agudo: eritema en palmas y plantas, edema de manos y pies
-
Subagudo: descamación periungueal de los dedos en la fase subaguda **
-
Exantema polimorfo
-
-
Inyección conjuntival bulbar bilateral sin exudado
-
Cambios en labios y cavidad oral :
-
Eritema o labios agrietados
-
Lengua aframbuesada
-
Eritema difuso de mucosa orofaríngea
-
-
Linfadenopatía cervical mayor de 1,5 cm de diámetro, normalmente unilateral
-
-
Exclusión de otras enfermedades con características similares.
* Pacientes con fiebre y menos de 4 criterios clínicos pueden ser diagnosticados de EK si se detectan alteraciones típicas en las arterias coronarias por ecocardiografía o arteriografía.
**Se ha propuesto incluir la descamación perineal como criterio.
Es característico que todas las manifestaciones clínicas no se presenten a la
vez en el tiempo, por lo que a veces es necesario esperar varios días antes de
hacer el diagnóstico. El término de Kawasaki incompleto se refiere a pacientes
que, aunque no cumplen suficientes criterios, pueden ser diagnosticados de EK.
Es más frecuente en menores de 1 año y mayores de 9. En 2004, la Asociación
Americana de Cardiología ha desarrollado un algoritmo de actuación para la EK
incompleta basada en opiniones de expertos que ayuda a usar los datos clínicos,
analíticos y ecocardiográficos para mejorar el diagnóstico y tratamiento
(Algoritmo 2). Debe considerarse el diagnóstico de EK en todos los niños con
fiebre inexplicable de 5 o más días de duración con 2 ó 3 criterios clínicos
principales y en cualquier niño menor de 6 meses con fiebre de más de 7 días de
duración con hallazgos de laboratorio compatibles con inflamación y sin causa
explicable de la fiebre(13).
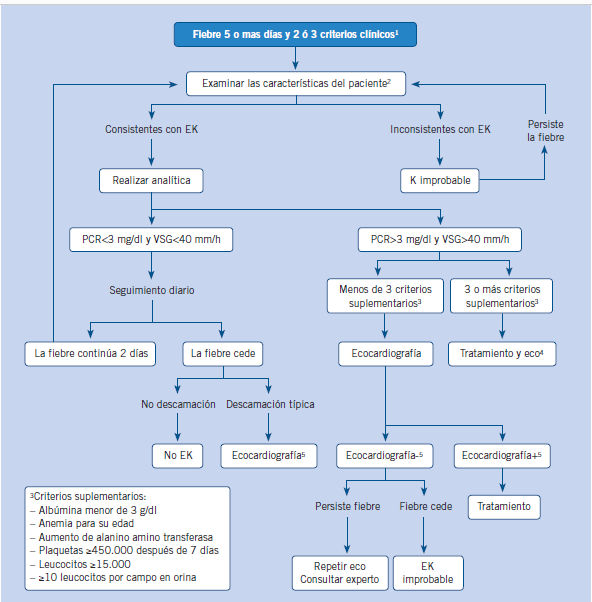
-
En los niños menores de 6 meses o fiebre de más de 7 días sin otra causa, se debe realizar una analítica y si se encuentran signos de inflamación, realizar una ecocardiografía, incluso si no cumple criterios clínicos.
-
Las características que sugieren otras enfermedades son conjuntivitis exudativa, faringitis exudativa, exantema vesicular o bulloso o adenopatías generalizadas.
-
Véase tabla.
-
Puede tratarse antes de la realización de ecocardiografía.
-
La ecocardiografía se considera positiva si se encuentra alguno de estos hallazgos: z score de arteria descendente anterior izquierda o arteria coronaria derecha ≥2,5, si cumplen los criterios de lesiones coronarias del Ministerio de Salud japonés (diámetro de la luz interna en niños menores de 5 años >3 mm y >4 mm en niños mayores de 5 años, cuando el diámetro interno de cualquier segmento mide al menos 1,5 veces lo que el segmento adyacente o cuando la luz arterial es claramente irregular), o existencia de ≥ de 3 características sugestivas que incluyen realce perivascular, ausencia de afilamiento, disminución de función del ventrículo izqdo., regurgitación mitral, derrame pericárdico o z score de la arteria descendente anterior izquierda o coronaria derecha 2-2,5.
Manifestaciones clínicas principales
La fiebre suele ser elevada y sin tratamiento persiste una media de 11 días. Los
cambios en las extremidades son característicos. En la fase aguda, suele
aparecer eritema de palmas y plantas. Pueden estar, además, edematosas y el niño
evita coger objetos y la deambulación. La descamación se inicia en la región
periungueal a las 2 ó 3 semanas del comienzo de la fiebre y puede extenderse a
palmas y plantas (Fig. 3).
A los 5 días del inicio de la fiebre, suele aparecer un rash eritematoso. Puede presentarse de muchas formas, aunque la más frecuente es una erupción maculopapular difusa inespecífica (Fig. 4). A veces, se manifiesta como: urticaria, rash escarlatiniforme, eritrodermia, similar al eritema multiforme o, menos frecuente, erupción micropustular. No se han descrito las formas bullosas, vesiculares y petequias. Suele localizarse en tronco y extremidades, acentuándose en la región perineal, donde puede aparecer una descamación temprana (Fig. 5).
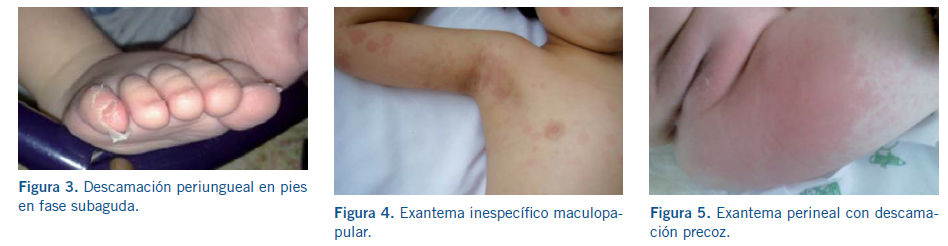
La inyección conjuntival bilateral suele aparecer poco después del inicio de la
fiebre. Afecta de forma característica a la conjuntiva bulbar y no se asocia a
exudado, edema conjuntival o ulceración corneal. Normalmente no es dolorosa. Los
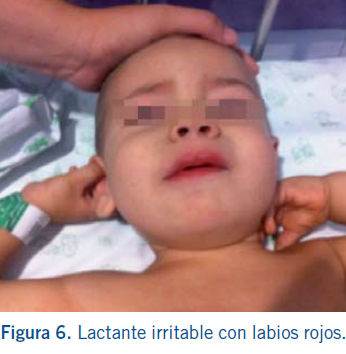
cambios en los labios y la cavidad oral incluyen: eritema (Fig.
6), sequedad, fisuras, descamación,
grietas y sangrado de labios, lengua aframbuesada indistinguible de la
escarlatina y eritema difuso de la mucosa orofaríngea. No suelen verse ni
úlceras orales ni exudado faríngeo.
La linfadenopatía cervical es la menos frecuente de todas las características clínicas principales. Suele ser unilateral y localizada en el triangulo cervical anterior. Para que sea criterio de la EK, debe haber una o más linfadenopatías mayores de 1,5 cm de diámetro. Suele ser firmes, no fluctuantes y sin eritema en la piel. Pueden aparecer otras manifestaciones descritas en la tabla VI (12,13).
Tabla VI. Manifestaciones clínicas y de laboratorio asociadas
-
Musculoesquelético
-
Artralgias o artritis: poliarticular grandes y pequeñas articulaciones
-
-
SNC
-
Irritabilidad : * meningitis aséptica
-
Parálisis facial periférica unilateral transitoria
-
Sordera neurosensorial transitoria
-
-
Afectación gastrointestinal
-
Diarrea, vómitos y dolor abdominal
-
Ictericia, hepatomegalia
-
Distensión acalculosa de vesícula biliar
-
-
Otras
-
Eritema e induración en sitio de vacunación de BCG
-
Uveítis anterior
-
Uretritis
-
Inflamación testicular
-
Nódulos e infiltrados pulmonares
-
Derrame pleural
-
-
Laboratorio
-
Leucocitosis con neutrofilia **
-
Anemia
-
Trombocitosis en fase subaguda (2º-4º semana)**
-
Aumento de PCR
-
Aumento de VSG
-
Alteración de lípidos
-
Hipoalbuminemia
-
Hiponatremia
-
Piuria estéril
-
Aumento de transaminasas
-
Pleocitosis en LCR
-
Leucocitosis en líquido sinovial
-
-
*Sobre todo en lactantes. Muy característico.
-
**La trombocitopenia y leucopenia son raras y pueden ser signos de asociación a síndrome de activación macrofágica.
Manifestaciones clínicas cardiovasculares asociadas
En los primeros 10 días, no suelen detectarse aneurismas coronarios, pero puede
apreciarse mediante ecocardiografía un aumento de la brillantez que rodea la luz
arterial o ectasias. Esta lesión precoz puede resolverse o evolucionar hacia
aneurismas. También, se puede observar disminución de la función ventricular,
regurgitación valvular o derrame pericárdico. Los aneurismas se suelen
detectar en la fase subaguda (4-6 semanas de enfermedad) (12,13).
Hallazgos de laboratorio
Las pruebas de laboratorio no son específicas, pero pueden contribuir al diagnóstico (Tabla VI). Suelen normalizarse de 6 a 10 semanas después del inicio de la enfermedad. El biomarcador N-terminal tipo pro-B péptido natriurético (NT-pro BNP) se correlaciona con marcadores de inflamación, estrés oxidativo y disfunción diastólica cardiaca. Algunos estudios sugieren que se eleva en pacientes con EK en comparación con controles febriles y que podría servir como dato analítico suplementario para el diagnóstico de EK incompleto, incluso podría ser marcador de riesgo de desarrollo de AC(12,13).
Diagnóstico diferencial
El polimorfismo de los signos y síntomas obliga a considerar un amplio diagnóstico diferencial que incluye procesos infecciosos, alérgicos o tóxicos y reumatológicos. El diagnóstico precoz, fundamentalmente de los casos atípicos, puede ser difícil.
-
Infecciones
-
Estreptocócicas y estafilocócicas mediadas por toxinas: en el caso de la escarlatina, existe exudado amigdalar, cultivo faríngeo positivo y respuesta al tratamiento con penicilina. En el shock tóxico, existe afectación renal, hipotensión y elevación de la creatinkinasa.
-
Sarampión: es típica la presencia de conjuntivitis supurativa, las lesiones de Koplik y la aparición del rash predominantemente en la cara.
-
Adenovirus: la conjuntivitis suele ser supurativa, y se puede utilizar para su diagnóstico los cultivos virales y la reacción en cadena de la polimerasa.
-
Otras: virus de Epstein-Barr, parvovirus B19, rickettsias, leptospiras.
-
-
Otros : Reacciones medicamentosas, artritis idiopática juvenil de comienzo sistémico, poliarteritis nodosa, lupus eritematoso sistémico, fiebre reumática, enfermedad del suero, Stevens-Johnson y síndromes autoinflamatorios (fiebre periódica asociada al receptor de TNF, síndrome hiper IgD, síndromes periódicos asociados a criopirinas)(12,13).
Tratamiento
El tratamiento con IGIV dentro de los 10
primeros días del comienzo de la enfermedad disminuye la incidencia de AC desde
un 20-25% a menos de un 5%.
Ya que no se ha podido establecer un score de riesgo que nos permita distinguir
a pacientes con mayor probabilidad de desarrollar aneurismas, todos los
pacientes diagnosticados de EK deben ser tratados con IGIV.
La aspirina se emplea por sus efectos antiinflamatorios (a dosis alta) y antitrombóticos (dosis bajas). Aunque no está demostrado que reduzca la incidencia de dilatación coronaria, sí parece disminuir la incidencia de infartos miocárdicos fatales. La dosis a emplear es controvertida, ya que no existen estudios de calidad. En la mayoría de los centros se suele emplear a dosis altas de 80-100 mg/kg/día en 4 dosis durante la fase aguda del proceso; mientras que, en Japón se emplea a dosis moderada (30-50 mg/kg/día). Cuando el paciente lleva 48-72 horas afebril, la dosis de aspirina se reduce a dosis antitrombóticas (3-5 mg/kg/día). El uso de ibuprofeno concomitante antagoniza el efecto antitrombótico de la aspirina. Los pacientes que reciben salicilatos de forma crónica deben vacunarse anualmente de la gripe y estar vacunados de la varicela(12,13).
La IGIV se utiliza a dosis elevadas (2 g/kg) en una sola infusión. Su
mecanismo de acción no es del todo conocido, parece tener un efecto
antiinflamatorio, disminuyendo la producción de citocinas proinflamatorias y
aumentando la producción de antagonista del receptor de IL1(17). Es un producto
seguro, con efectos secundarios poco frecuentes :
reacción infusional, anafilaxia, urticaria, meningitis aséptica, anemia
hemolítica autoinmune, tromboembolismo y fallo renal agudo. Aunque sólo se
han demostrado los beneficios para pacientes tratados en los primeros 10 días de
la enfermedad, algunos autores recomiendan su empleo en niños con evidencia de
inflamación persistente, con o sin alteraciones coronarias, que son
diagnosticados después de esta fecha. Una vez administrada, deben esperarse 11
meses para la vacunación de virus vivos.
La utilización de corticoides es controvertida. En un metaanálisis reciente, concluyen que la combinación de corticoides con el régimen convencional de IGIV como tratamiento inicial reduciría el riego de desarrollo de aneurismas coronarios sin aumentar los efectos secundarios( 18,19).
Tratamiento de la enfermedad de Kawasaki refractaria
Entre 11,6 y 38,3% de pacientes tratados inicialmente con IGIV y aspirina a
dosis alta, tienen fiebre persistente 24- 48 horas tras la primera dosis de IGIV
o bien recurre a las 36 horas o más después de completar la infusión de IGIV.
Estos pacientes tienen mayor riesgo de desarrollo de AC. La mayoría de los
expertos recomiendan retratamiento con IGIV a 2 g/kg, previa reevaluación
clínica por si el diagnóstico inicial cambia. En algunos estudios, se ha
señalado que los pacientes retratados que reciben la 2ª dosis de IGIV
precozmente (antes de los 10 días del inicio de la enfermedad) tienen menor
prevalencia de aneurismas coronarios. Recientemente, se ha publicado que el
tratamiento con infliximab (anticuerpo monoclonal anti TNF-α) puede ser tan
seguro y efectivo como la IGIV en EK refractaria en cuanto a duración de
síntomas; si bien, en estudios retrospectivos no se han encontrado diferencias
en cuanto a la aparición de aneurismas entre el grupo de infliximab y el de IGIV(20).
En caso de fracaso de la 2ª dosis de IGIV, se pueden considerar pulsos de
metilprednisolona (30 mg/kg durante 3 días), ciclofosfamida asociada a
prednisona, ciclosporina o plasmaféresis.
Existen en la actualidad nuevos tratamientos de utilidad aún incierta por la
falta de estudios, como la ulinastatina (inhibidor de la tripsina humana) que se
ha probado en EK refractario a IGIV, el abciximab (anticuerpo monoclonal
trombolítico), que se ha empleado en pacientes con aneurismas gigantes en la
fase subaguda, y atorvastin, estatina estudiada en ratones, que podría detener
la inflamación en EK(13).
Pronóstico
Muchos casos de infartos en personas
jóvenes (3ª-4ª década) son atribuidos a una infradiagnosticada EK en la
infancia.
La mortalidad (0,17% en Estados Unidos) se relaciona siempre con secuelas
cardiacas. La causa más frecuente de muerte es el infarto de miocardio por
trombosis de aneurismas, que suele suceder durante el primer año de enfermedad(
13). La severidad de la anemia, trombocitopenia, hipoalbuminemia y elevación
de alanino aminotransferasa (ALT) por encima de 200 U/L y PCR y VSG muy elevadas
o persistentemente aumentadas se correlacionan con el riesgo de
desarrollar AC. También, son factores de riesgo de aparición de AC las edades
extremas y la larga duración de la fiebre antes del tratamiento (menores de 6
meses y mayores de 8 años suelen tener una presentación atípica, una mayor
demora diagnóstica y probablemente a una mayor vulnerabilidad genética)(12).
En Japón se han desarrollado varios score
de riesgo para predecir la resistencia a IGIV y poder tratar a estos pacientes
de forma más agresiva (podrían beneficiarse de un tratamiento inicial con
corticoides). Estos score no son aplicables a niños de Estados Unidos(12). Un 7%
de EK se presenta como shock, y se suele asociar con disfunción ventricular y
refractariedad al tratamiento.
El 1,9% de pacientes con EK desarrollaron un síndrome de activación macrofágica,
manifestándose como fiebre resistente a IGIV y aumento de transaminasas.
El mejor pronóstico se asocia a los aneurismas fusiformes de menos de 8 mm de
diámetro y el peor con los aneurismas gigantes (>8 mm de diámetro). En la mitad
de los pacientes con aneurismas coronarios, estos regresan a largo plazo y en un
20% de los casos progresan hacia una estenosis coronaria. El seguimiento
evolutivo de pacientes con EK se basa en la estratificación de riesgo relativo
de isquemia miocárdica.
Se suele recomendar estudio ecocardiográfico al diagnóstico, a las 2 y a las 6-8
semanas del comienzo de la enfermedad, y en función de los hallazgos, se
establecen los niveles de riesgo :
-
Nivel de riesgo I: pacientes sin AC en ninguno de los estadios. Pueden suspender tratamiento con aspirina a las 6-8 semanas de inicio de la enfermedad.
-
Nivel de riesgo II: pacientes con ectasia transitoria de la arteria coronaria en la ecocardiografía (desaparece durante la enfermedad aguda) que pueden recibir recomendaciones similares a las del nivel I y continuar reevaluación cardiológica cada 3-5 años.
-
Nivel de riesgo III, IV y V: pacientes con aneurismas que precisan mantener tratamiento con: aspirina, con anticoagulación si son aneurismas gigantes, restricción de actividad física y controles cardiológicos periódicos.
La resonancia magnética angiográfica
coronaria y la tomografía computada multicorte son herramientas no invasivas que
en el futuro pueden reemplazar a la angiografía en el seguimiento de pacientes
con aneurismas coronarios(13).
El riesgo cardiovascular a largo plazo en pacientes con EK sin AC es
desconocido; si bien, parece que puede ser un factor de riesgo para el
desarrollo de arteriosclerosis precoz en la edad adulta debido a una disfunción
endotelial, por lo que se deben generalizar a todos ellos las recomendaciones
sobre factores de riesgo cardiovascular: dieta saludable, ejercicio moderado,
peso adecuado, control de tensión arterial y evitar consumo y exposición al
tabaco.
Panarteritis nodosa infantil
(PAN)
Es una enfermedad rara en niños de etiología desconocida, aunque se han
implicado el virus de la hepatitis B, la infección estreptocócica y los
depósitos de inmunocomplejos. Se ha asociado con la fiebre mediterránea
familiar. Se puede diagnosticar a un niño de PAN si en la biopsia presenta
vasculitis necrotizante de arterias de pequeño y mediano calibre o angiografía
patológica y al menos 1 de los siguientes: afectación de piel (lívedo
reticularis, nódulos, infartos…), mialgias o debilidad muscular, hipertensión
arterial en relación con su edad, mononeuropatía o polineuropatía o afectación
renal (hematuria, proteinuria o afectación de la función renal)(1,2,7).
Poliarteritis cutánea
Es más frecuente en niños y se caracteriza por la presencia de nódulos
subcutáneos dolorosos, lesiones no purpúricas, con o sin lívedo reticularis, sin
afectación sistémica (excepto mialgia, atralgia y artritis no erosiva). Se
asocia a menudo con antecedente de infección estreptocócica(1,2,7).
Vasculitis asociada a ANCA
Son vasculitis de mediano y pequeño vaso con elevada morbi-mortalidad y
frecuentes recaídas. La granulomatosis de Wegener afecta al tracto respiratorio
superior e inferior y riñones. 90% son ANCA positivos. Para diagnosticarla deben
estar presentes al menos 3 de las 6 características: afectación renal
(hematuria, proteinuria, glomerulonefritis pauciinmune necrotizante), granulomas
necrotizantes en las biopsias, afectación de vía aérea superior, afectación
laringotraqueobronquial, radiografía o TAC pulmonares patológicos y ANCA
positivos.
La poliangeítis microscópica es una enfermedad no granulomatosa necrotizante que
afecta a vasos pequeños. Es característica la capilaritis pulmonar y
glomerulonefritis necrotizante. Casi el 100% tienen afectación renal y se asocia
con títulos elevados de ANCA. El Churg- Strauss es una vasculitis granulomatosa
de vaso mediano y pequeño que afecta a pacientes con asma severa o
alergia(1,2,7).
Arteritis de Takayasu
Vasculitis granulomatosa que afecta de forma primaria a los grandes vasos como
la aorta y sus ramas. Se presenta en mujeres entre 10 y 30 años, con una
incidencia mayor en asiáticos. Para su diagnóstico es necesaria anormalidad en
arteriografía (convencional, angioRNM o angio TAC) de la aorta y sus ramas
principales, más al menos una de las siguientes características :
disminución del pulso arterial periférico y/o claudicación de extremidades,
diferencia de presión mayor de 10 mmHg en cualquier miembro, soplo en la
aorta o sus ramas principales, hipertensión en relación con su edad, aumento de
reactantes de fase aguda 1,2,7).
Vasculitis primaria del
sistema nervioso central
La incidencia en niños es desconocida, debido a lo poco frecuentes que son y a
la carencia de un consenso para los criterios diagnósticos. Los criterios
para adultos son: déficit neurológico con hallazgos típicos de vasculitis en la
angiografía o histología. Lo más frecuente es que se manifieste como cefaleas y
déficits neurológicos focales, aunque también aparecen con frecuencia
disfunciones cognitivas y cambios de comportamiento. El papel de las diversas
técnicas de neuroimagen en el diagnóstico y seguimiento de esta patología no
está plenamente establecido. La biopsia craneal es la prueba oro, pero tiene una
sensibilidad limitada debido a la naturaleza desigual de la enfermedad(1,2,7).