Casos clínicos
Febrero 2015
BMJ Case Reports 2015
Colaboración Dra Alexis Strickler
1 ) Caso 1 : Postoperative epidural haematomas associated with hydrocephalus caused by intraoperative overdrainage of cerebrospinal fluid: two case reports with a literature review of 19 cases
Manabu Niimura y cols , Department of Neurosurgery, Tokyo Metropolitan Neurological Hospital, Tokyo, Japan
Summary
We report two cases with postoperative epidural haematomas (EDHs) associated with hydrocephalus and discuss the cause of haematoma development on the basis of a literature review. A 13-year-old boy presented with obstructive hydrocephalus caused by a sellar mass lesion. Multifocal EDHs occurred after partial resection of the lesion via a transcallosal approach following ventricular drainage. In the second case, a 26-year-old man who had a history of ventriculoperitoneal shunting for congenital hydrocephalus presented with hydrocephalus caused by ventricular catheter obstruction. An EDH occurred after replacement of the ventricular catheter with a new burr hole opening. On the basis of a review of 19 cases including our two cases, the authors concluded that postoperative EDH development associated with hydrocephalus was mostly caused by intraoperative overdrainage of cerebrospinal fluid, resulting in rapid shrinkage of the brain with dilation of the epidural space, a situation that may have caused dural venous bleeding.
2) Caso 2 : A case of lissencephaly in a 5-month-old infant
Adeel Ejaz Syed , Department of Paediatrics, Princess Alexandra Hospital, Essex, UK
Description
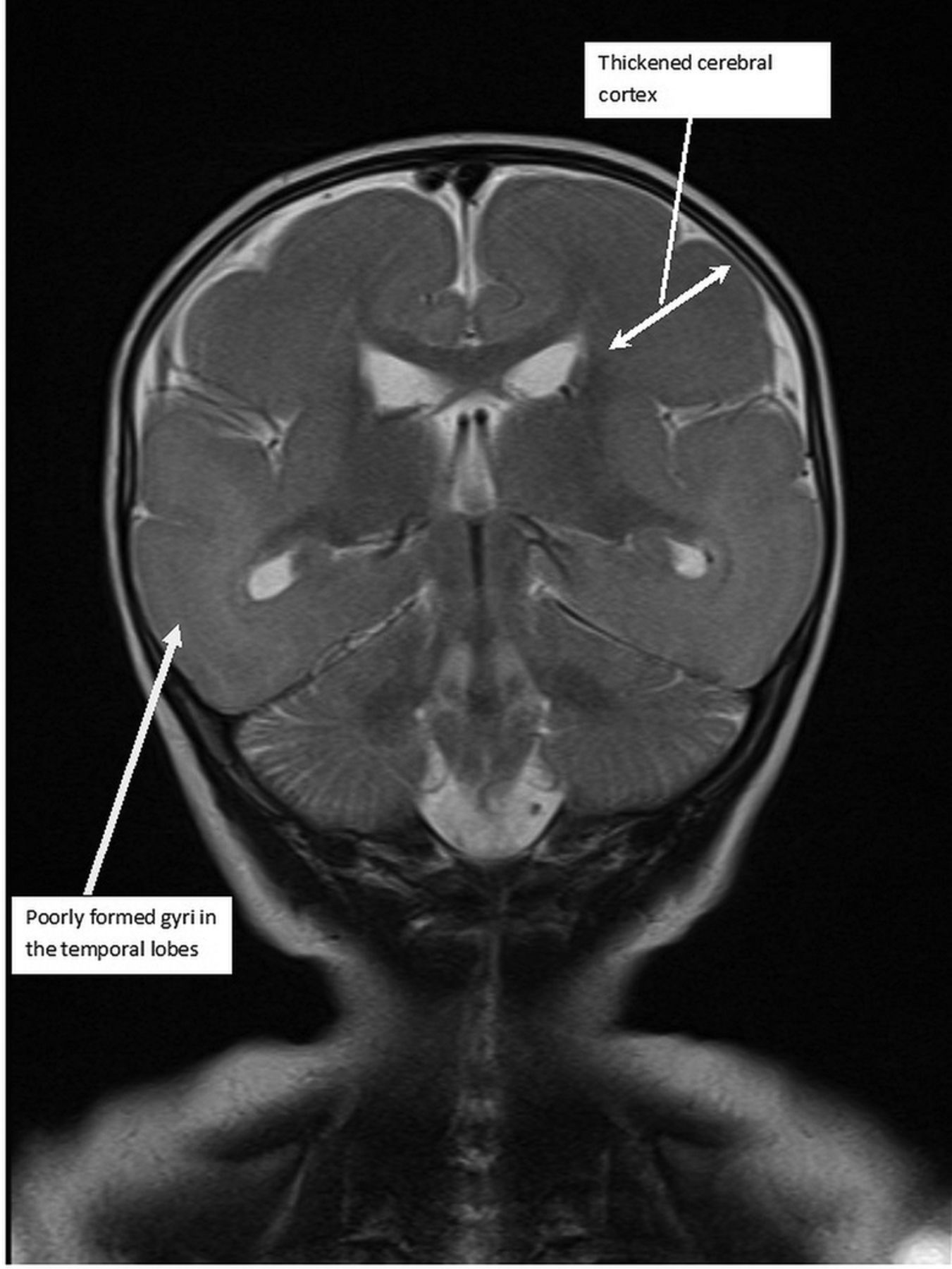
A 5-month-old full-term male patient, born of a non-consanguineous marriage presented to A&E department following parental concerns about abnormal movements and developmental regression. Development halted at 4 months of age. The patient increasingly showed episodes with flickering of eyes and tonic movements followed by postictal behaviour. He stopped fixing and following, stopped responding to loud noises, stopped reaching out for objects, and had stopped babbling or making any noises, all of which he had been doing previously. Examination revealed a child with a frog-like posture who appeared very floppy, and with a relatively small head circumference of 41.7 cm (9th centile). Initial blood examinations revealed no significant abnormality. An EEG indicated hypsarrhythmia and a brain MRI was performed. This showed :
Very few sulci with thick cortex;
Some gyri formation along frontal and temporal lobes (pachygyria);
Circumferential band of high signal change in parieto-occipital cortex.
This was all consistent with lissencephaly type 1, and genetic tests later confirmed a corresponding LIS1 c. 1050delG mutation. Owing to poor effect with other antiepileptics, Vigabatrin was started in this patient to control his multiple seizures.
Lissencephaly means smooth brain, and is caused by a neuronal migration defect1 resulting in a severe lack of gyrus and sulcus formation. LIS1-associated lissencephaly includes Miller-Dieker syndrome, isolated lissencephaly sequence (ILS) and others,2 which can be established by using fluorescence in situ hybridisation. Prognosis is relatively poor with most children passing away by age 10. Figures 1⇓–3 show the various MRIs and their sequences.
Learning points
Lissencephaly (smooth brain) represents a group of disorders with neuronal migration defects that are characterised by severely reduced gyral and sulcal formation (aygria or pachygyria).
Lissencephaly has no cure, and management is largely via supportive treatment.
Figure 1 : T2-weighted coronal image showing poorly formed gyri in the temporal lobes and a thickened cerebral cortex.
View larger version: In a new window
3) Caso 3 : Pneumatosis intestinalis presenting as pneumoperitoneum in a teenage girl with pyloric stenosis
C W Y Wong y cols. , Division of Paediatric Surgery, Department of Surgery, Queen Mary Hospital, The University of Hong Kong, Hong Kong
Summary
A 16-year-old girl presented with free gas under the diaphragm after endoscopic balloon dilation for pyloric stenosis. There was no perforation site identified on laparotomy. However, there was massive pneumatosis intestinalis involving the entire small bowel.
4) Caso 4 : Use of a medication passport in a disabled child seen across many care settings
Barry Jubraj y cols , Department of Pharmacy, Chelsea & Westminster Hospital NHS Foundation Trust, London, UK
Summary
Written information for patients about their medicines has demonstrable benefits for their understanding and adherence. In the UK, no single, complete record of medications for individual patients can be guaranteed. Therefore, patients and carers are often relied on to recall the complete medication list, which can be a challenge given multiple and potentially stressful appointments. Wide-ranging feedback suggests that a medication ‘passport’ developed by the National Institute for Health Research Collaboration for Leadership in Applied Health Research and Care North West London (NIHR CLAHRC NWL) has benefited elderly patients, who often attend many appointments where the current medication list may not be available. We describe the use of this passport (known as ‘My Medication Passport’—MMP) in a child with multiple disabilities. The practical advantages are explored, including the potential for a paediatric version to facilitate discussions around the administration of medicines. MMP is an early example of a useful tool to help children and young people, parents and carers to manage medicines more effectively.
5) Caso 5 : Dental manifestations of congenital rubella syndrome
Ruchi Ahuja y cols, Department of Pedodontics and Preventive Dentistry, Peoples’ College of Dental Sciences and Research Centre, Bhopal, Madhya Pradesh, India
Description
Congenital rubella syndrome (CRS) has a low incidence and has been expected to reduce to 1/100 000 live-births. The infection can be acquired by contact with the togavirus family of microorganisms during pregnancy. Fetal development is completely paralysed once the microorganism is transmitted to the fetus.
Patients with CRS are usually categorised as special patients with exaggerated dental problems usually due to lacking manual dexterity and failure on the part of caretakers to provide timely oral disease prevention as they consider it secondary owing to the grave systemic problems the patient suffers from. Treatment cannot be performed in a routine setup but requires physical restraints or sedation to cope with the patient's physical and intellectual limitations.
In the present case, a 4-year-old girl had CRS with severe hearing disorders, congenital cardiopathy (ventricular septal defect, VSD) for which she had undergone surgical intervention 1-year prior. She had a minimal attention span and poor ability to respond to instructions given. Her mother revealed that as the child was unable to chew, her diet usually comprised of soft foods. Intraoral examination revealed not all teeth were cariously involved, however, the lower anterior teeth were diagnosed congenitally missing owing to a knife edged alveolar ridge (figure 1). Also, the maxillary arch was unusually narrow and the palate was deep (figure 2). Symptomatic relief of pain was provided by excavation and temporisation in decayed teeth. Comprehensive treatment could not be planned under general anaesthesia due to the unwillingness of the parents for the procedure.
Learning points
Congenital rubella syndrome (CRS) can be acquired by contact with the togavirus family of microorganisms during pregnancy.
Patients with CRS are usually categorised as special patients with exaggerated dental problems usually due to lacking manual dexterity.
Treatment cannot be performed in a routine setup but requires physical restraints or sedation to cope with the patient's physical and intellectual limitations.
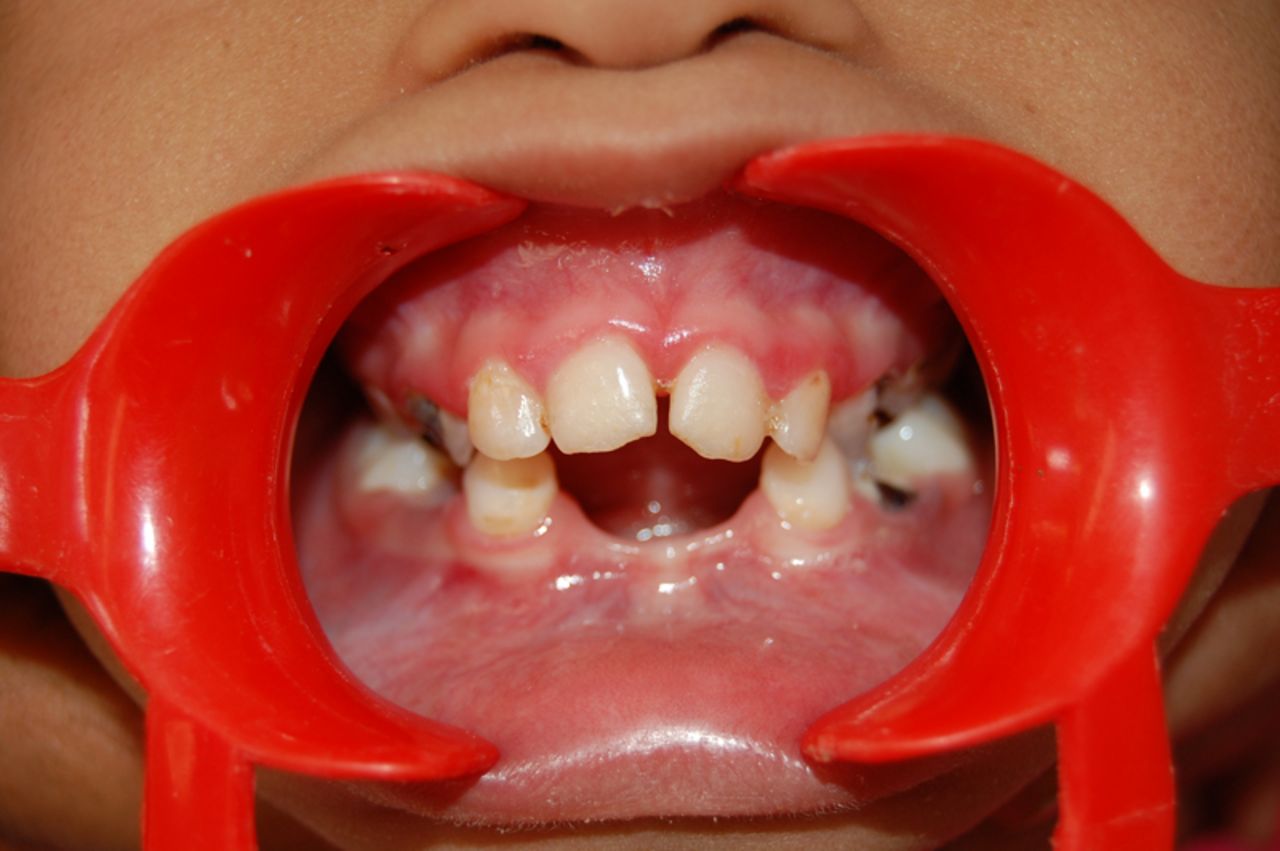
Figure 1 Congenitally missing lower incisors.
View larger version: In a new window

Figure 2 : Constricted maxillary arch and high palate.
View larger version: In a new window
6) Caso 6 : Antenatal diagnosis of intracranial haemorrhage and porencephalic cyst
T Williams y cols , Simpson Centre for Reproductive Health, Royal Infirmary of Edinburgh, Edinburgh, UK
Description
A 27-year-old woman was referred at 37 weeks’ gestation for confirmation of malpresentation. Incidental hydrocephalus was found on ultrasound. Fetal MRI confirmed dilation of the lateral and third ventricles and an extensive mixed signal intensity cystic structure in the left hemisphere (7×6×4 cm) with evidence of blood products and restricted diffusion (figure 1A). The maternal platelet count was 114×109/L, and maternal antibody testing for neonatal alloimmune thrombocytopaenia (NAIT) was positive (anti-HPA (human platelet-specific antigen) 1a alloantibodies, maternal genotype HPA 1bb, paternal genotype HPA 1ab).

Figure 1 : (A) Antenatal MRI. (B) Postnatal MRI day 1.
View larger version : In a new window
Subsequently a live female infant was delivered by caesarean section. At birth the infant had widened sutures and a petechial rash. The platelet count was 4×109/L and the infant received two HPA1a-negative platelet transfusions shortly after birth. MRI the next day confirmed posthaemorrhagic ventricular dilation and a large haemorrhagic parenchymal infarction with porencephalic cyst (figure 1B, arrow). She required a cystoventriculoperitoneal shunt at 4 weeks.
NAIT is an IgG mediated disorder that occurs after maternal exposure to incompatible fetal paternally derived platelet antigens; it can occur in first pregnancies. Outcomes for NAIT with intracranial haemorrhage (ICH) are poor: registry data suggest that only 12% of children with ICH secondary to NAIT survive without significant neurodisability. Detailed antenatal imaging and testing for NAIT when fetal intracranial haemorrhage is suspected enables planned early treatment with matched platelets if the diagnosis is confirmed, which minimises the duration of postnatal profound thrombocytopenia and risk of further haemorrhage.